
La armonización de las buenas prácticas y los requisitos regulatorios, permitirán a los desarrolladores encontrar el mejor enfoque de pruebas de estabilidad.
Los laboratorios de calibración tambien conocidos como laboratorios de metrología son un pilar para nuestra industria.
Dentro de la industria bio/farmacéutica, determinar la estabilidad del producto farmacéutico principio activo es una parte integral del proceso de desarrollo y debe realizarse de acuerdo con los requisitos regulatorios específicos. Deben emplearse métodos estrictos de prueba para determinar la estabilidad, la seguridad, la integridad y la vida útil de un producto en una variedad de condiciones estipuladas.
Las pruebas minuciosas de estabilidad se consideran un requisito previo para la aceptación o aprobación de cualquier producto bio/farmacéutico por parte del organismo regulador pertinente. “La evaluación de la estabilidad es potencialmente el aspecto más importante del desarrollo de productos farmacéuticos,” dice Stuart Kirbyshire, gerente de estabilidad en Intertek harmaceutical Services. “Cualquier autoridad regulatoria esperará que usted tenga un conocimiento completo y profundo de su producto, y será problemático obtener la aprobación sin comprender los cambios en la estabilidad química y física bajo variaciones de temperatura, humedad y luz.”
Las normativas, proporcionadas por la Conferencia Internacional de Armonización (ICH, por sus siglas en inglés), la Organización Mundial de la Salud (OMS) y otras agencias, especifican que los productos bio/farmacéuticos deben tener una fecha de caducidad determinada a través de protocolos de prueba de estabilidad más apropiados. Estas normativas tienen en cuenta la zona climática del mercado previsto.
“También es clave evaluar su producto en una amplia gama de condiciones climáticas para garantizar la llegada al mercado global,” confirma Kirbyshire.”También es deseable comprender los efectos del transporte y los ciclos de temperatura, para demostrar que no habrá eventos imprevistos durante el envío y el almacenamiento.”
Sin embargo, algunas de las normativas proporcionadas para las pruebas de estabilidad solo son aplicables a productos farmacéuticos/principios activos convencionales de moléculas pequeñas y, como tales, no proporcionan metodologías específicas para las pruebas recomendadas. Por lo tanto, los analistas deben desarrollar métodos analíticos y bioensayos que luego deben validarse.
“Las técnicas analíticas empleadas pueden variar según el tipo de molécula, la forma farmacéutica y el uso terapéutico previsto,” explica Kirbyshire. “El objetivo principal para realizar estudios de estabilidad, es determinar la vida útil, la caracterización de los productos de degradación y la potencia de cualquier producto farmacéutico.”
“Para productos biológicos, debido a su complejidad estructural, dentro de la industria bio/farmacéutica, determinar la estabilidad del producto farmacéutico principio activo es una parte integral del proceso de desarrollo y debe realizarse de acuerdo con los requisitos regulatorios específicos. Deben emplearse métodos estrictos de prueba para determinar la estabilidad, la seguridad, la integridad y la vida útil de un producto en una variedad de condiciones estipuladas.
Las pruebas minuciosas de estabilidad se consideran un requisito previo para la aceptación o aprobación de cualquier producto bio/farmacéutico por parte del organismo regulador pertinente. “La evaluación de la estabilidad es potencialmente el aspecto más importante del desarrollo de productos farmacéuticos,” dice Stuart Kirbyshire, gerente de estabilidad en Intertek Pharmaceutical Services. “Cualquier autoridad regulatoria esperará que usted tenga un conocimiento completo y profundo de su producto, y será problemático obtener la aprobación sin comprender los cambios en la estabilidad química y física bajo variaciones de temperatura, humedad y luz.”
Las normativas, proporcionadas por la Conferencia Internacional de Armonización (ICH, por sus siglas en inglés), la Organización Mundial de la Salud (OMS) y otras agencias, especifican que los productos bio/farmacéuticos deben tener una fecha de caducidad determinada a través de protocolos de prueba de estabilidad se requiere una capacidad analítica más diversa para un estudio de estabilidad. Por lo tanto, es importante diseñar programas de estabilidad que incorporen estrategias analíticas robustos,” señala Jordi Trafach, director asociado de Biologics, Intertek Pharmaceutical Services. “Estos enfoques analíticos deben basarse en una buena comprensión científica de las vías de degradación y los métodos establecidos indicadores de estabilidad, los cuales son específicos y sensibles con respecto a los cambios en los atributos críticos de calidad (CQA, por sus siglas en inglés).”
La calibración de potenciómetros así como instrumentos de presión son aspectos clave relacionados con productos biológicos
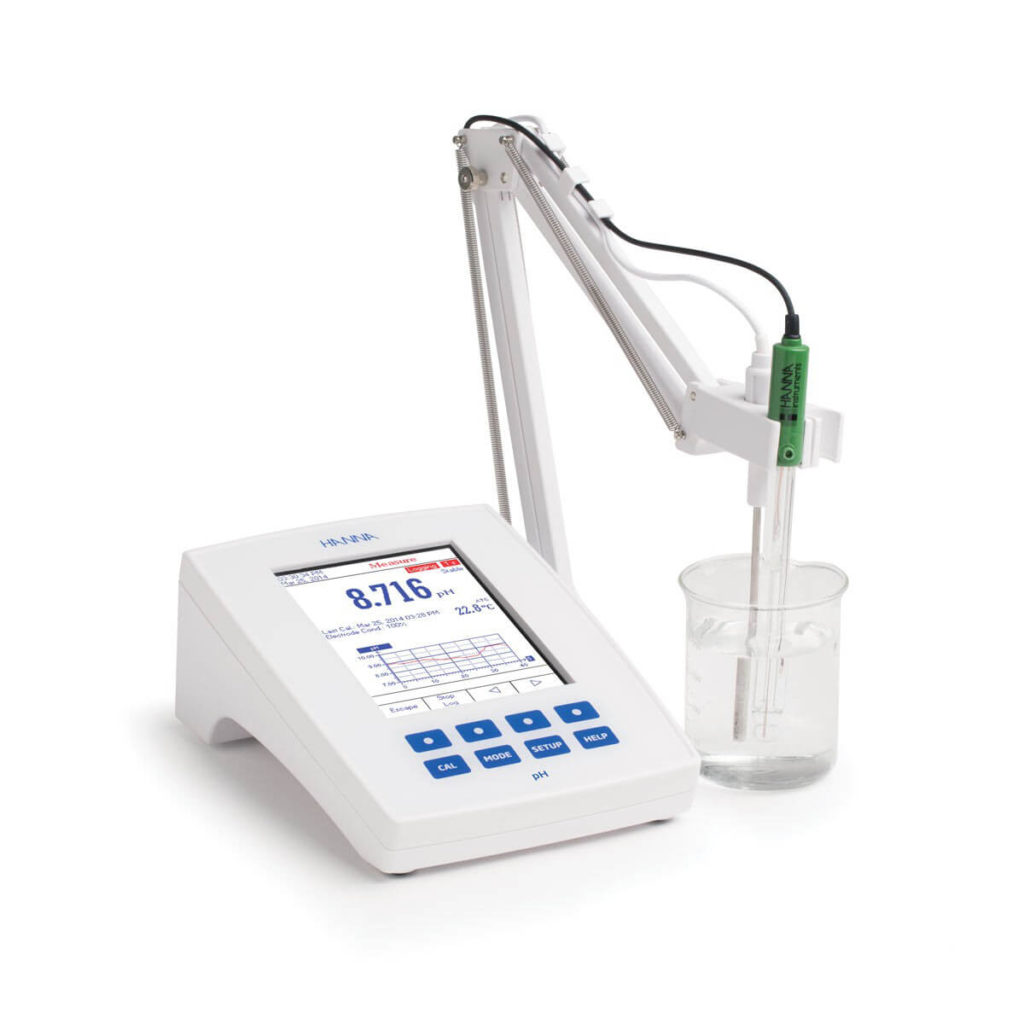
Se sabe que los productos biológicos son extremadamente sensibles a factores ambientales y susceptibles de agregarse y degradarse, ese es el motivo por el cual la industria farmacéutica debe recurrir a expertos en la calibración de instrumentos para que se pueda verificar que existen condiciones ambientales estables durante la elaboración de biológicos y reactivos así como en el correcto almacenamiento y distribución. El servicio de calibración de potenciómetro o medidor de pH nos ayuda mucho durante la fabricación de medicamentos y materias primas. “Para reducir el riesgo de degradación y mantener la actividad biológica del producto, se deben establecer las condiciones adecuadas para el almacenamiento y la vida útil,” enfatiza Trafach. “La comprensión de las posibles rutas de degradación en relación con el entorno de almacenamiento es fundamental para establecer qué CQAs son más susceptibles de cambiar a lo largo de la vida del agente terapéutico. En última instancia, esta comprensión garantiza que se implemente la estrategia óptima de control de calidad para monitorear la eficacia y seguridad continuas de un agente terapéutico”.
ICH es la Conferencia Internacional de Armonización. RMN es resonancia magnética nuclear. FTIR es espectroscopia infrarroja por transformada de Fourier. HPLC es cromatografía líquida de alta resolución. CG es cromatografía de gases. UV es ultravioleta. IC es cromatografía de iónica. ICP es plasma de acoplamiento inductivo. CE es electroforesis capilar. SOS es dodecil sulfato de sodio. CL es cromatografía líquida. iCIEF es imágenes de enfoque isoeléctrico capilar.
Como resultado de la naturaleza compleja de los productos biológicos, Trafach afirma que se pueden requerir métodos no comerciales no farmacopéicos para monitorear todos los productos de degradación potenciales y conocidos a lo largo del desarrollo del producto. Destaca que también es importante tener en cuenta los excipientes que frecuentemente se requieren en las formulaciones biológicas, ya que estos excipientes pueden ser susceptibles a la degradación o también pueden reaccionar con el producto biológico principal.
“En los casos en los que se observa un potencial producto de degradación en una etapa de desarrollo posterior, puede ser necesario volver a realizar la prueba, lo que podría demorar la finalización del estudio, potencialmente impactando el registro del producto,” continúa Trafach. “Los métodos indicadores de estabilidad deben optimizarse y validarse para demostrar precisión, exactitud y robustez y que el desempeño del método es adecuado para el uso previsto.”
Estudios de degradación forzada
Los estudios de degradación forzada se utilizan para determinar productos de degradación que se forman durante los estudios farmacéuticos de estabilidad acelerada y de estabilidad a largo plazo. Estos estudios son estipulados por los organismos reguladores para nuevos principios activos y son parte integral para comprender las vías de degradación y, como tales, obtener una evaluación completa de la estabilidad biológica, señala Trafach.
“Los estudios de degradación forzada también generan información invaluable sobre el desarrollo de la formulación y el desarrollo del proceso,” agrega. “Un estudio de degradación forzada puede incluir factores como temperatura, luz, pH, agentes oxidantes, estrés mecánico y ciclos de congelación-descongelación. El conocimiento de estos estudios, junto con una comprensión detallada del producto y el proceso, ayudan a establecer los CQA.”
Planear es prudente, el uso de manómetros es fundamental

El nivel de complejidad y la experiencia científica necesaria para realizar pruebas de estabilidad de calidad y eficiencia suficientes, puede llevar un período de tiempo considerable e incurrir en costos significativos. Sin embargo, la planificación adecuada del procedimiento puede ser útil, según Kirbyshire. La calibración de manómetros e instrumentos de presión han sido servicios especializados relevantes, importantes aliados de los cuales no podemos prescindir, han ayudado de manera muy positiva a la industria farmacéutica para controlar procesos de producción de diversos medicamentos. “Las presiones de tiempo pueden facilitarse con una buena planificación de otras actividades relacionadas con los estudios a largo plazo, respaldada por el diseño de análisis de extremos y de matriz de lotes, los tipos de envase y las fortalezas, como se detalla en ICH QlD,” dice.
“Además de la planificación,” continúa Kirbyshire, “las pruebas de un producto almacenado en condiciones de estrés pueden proporcionar una indicación temprana de cómo la estabilidad a largo plazo puede resultar en el futuro.”
En el futuro, Kirbyshire cree que habrá un mayor enfoque en los beneficios de los programas de evaluación de estabilidad acelerada (PEEA) durante el desarrollo de la etapa inicial, los cuales tienen el potencial de expandirse a evaluaciones de la etapa posterior. “La estrategia PEEA se basa en métodos robustos para la degradación forzada, donde los productos se someten a condiciones de elevada temperatura y humedad, y los plazos de tiempo se reducen a 14 días, los cuales se pueden usar para predecir posibles efectos de estabilidad a largo plazo,” explica.
Sin embargo, a pesar de que se ha observado cierto éxito en el campo de las moléculas pequeñas, Kirbyshire subraya que la idoneidad para las pruebas de estabilidad física o la evaluación de moléculas grandes no están actualmente probadas. “Con un mayor uso de la técnica, se harán más avances al establecer PEEA como un método confiable y rentable para determinar la estabilidad del producto,” dice.
Teniendo en cuenta el producto terminado
La estabilidad de un producto bio/farmacéutico dependerá de la forma farmacéutica y del sistema de contenedor cierre elegido para la implementación comercial. “La vida útil calculada y el almacenamiento comercial de su producto, una vez que esté disponible comercialmente, influirá fuertemente en la estrategia de las pruebas de estabilidad durante el desarrollo,” confirma Kirbyshire.
Por ejemplo, las soluciones parenterales pueden tener una mayor sensibilidad a extremos de temperatura y luz, lo cual normalmente conduce a un rango objetivo de temperatura de almacenamiento de 2-8ºC, explica. “Por lo tanto, 12 meses de almacenamiento a 2-8ºC y seis meses de estabilidad acelerada a 25ºC/60% de humedad relativa (HR) son suficientes para establecer la vida útil,” señala.
Kirbyshire enfatiza que en los casos de inhaladores de polvo seco (DPI, por sus siglas en inglés) y formas farmacéuticas sólidas orales, la humedad es el atributo clave a evaluar. “Se requieren estudios a largo plazo en condiciones de humedad elevada (p. ej., 30ºC/75% de HR y 40ºC/75% de HR),” dice. “Una prueba crítica en todo momento es la evaluación de la distribución aerodinámica del tamaño de partícula para los DPI y la disolución para las formas farmacéuticas sólidas orales, lo cual confirmará la eficacia continua. Cualquier reducción en el desempeño puede mejorarse mediante el uso de un empaque efectivo y, potencialmente, la adición de materiales desecantes. Los estudios de estabilidad son claves para determinarlos.”
Pruebas de estabilidad subcontratadas
Aquí es en donde los servicios de calibración de laboratorios de calibración acreditados ante ema toman gran relevancia, acercarse con un laboratorio de calibración sin lugar a dudas ha aumentado la calidad de los productos y ha echo a la industria mas competitiva.

Laboratorio de calibración presión 
Calibración de manómetros
Nos ha permitido contar con la calidad requerida por los diferentes laboratorios y mercados a lo largo del globo.
La decisión de subcontratar estudios de estabilidad también proveídos por laboratorios de calibración acreditados puede tener un impacto significativo, ya sea positivo o negativo, en el éxito del programa general de desarrollo de medicamentos. Si no se elige un socio con la debida atención, la decisión del patrocinador podría resultar costosa.
”Al evaluar a cualquier socio de estabilidad subcontratada, hay muchas cosas que considerar,” revela Kirbyshire. “Deben tener un conocimiento profundo de la normativa regulatoria clave, ICH QlA R2, pero también deben demostrar el cumplimiento con [Organización Internacional de Normalización] ISO 9001 para proporcionar plena confianza en los sistemas de calidad y la integridad de datos.”
Otros aspectos que se deben tener en cuenta al elegir un socio subcontratado son los programas de calificación y mantenimiento de las instalaciones de almacenamiento y los sistemas de monitoreo asociados teniendo cuidado que sea un laboratorio acreditado ante ema Entidad Mexicana de Acreditación, continúa Kirbyshire. “La integridad del estudio es tan buena como la confiabilidad y la robustez de los equipos y las estrategias de control utilizadas por el proveedor,” agrega. “Las desviaciones de las condiciones de almacenamiento pueden tener un efecto impredecible y gravemente perjudicial para cualquier estudio, desde una evaluaci6n a corto plazo de la estabilidad del API, hasta un registro completo del programa de estabilidad que se extiende hasta por cinco afros.”
Mejoras significativas
Según Kirbyshire, a lo largo de los años ha habido mejoras significativas en la confiabilidad de los equipos y los sistemas de monitoreo, incluyendo la retroalimentación en tiempo real de desviaciones potenciales. Estas mejoras tecnológicas han dado lugar a decisiones rápidas y eficientes que deben tomarse con respecto a la siguiente etapa del desarrollo del producto. “Las decisiones regulatorias están cambiando de manera rutinaria según los datos disponibles actualmente y las estrategias justificadas de evaluación de estabilidad,” resume. “A través de la armonización de esta información, las compañías que desarrollan medicamentos pueden tomar decisiones informadas sobre la mejor estrategia a adoptar para sus productos”.